Rhumatologie
Maladie de Behçet : des tableaux polymorphes et un traitement individualisé
La maladie de Behçet représente un défi médical complexe en raison de ses manifestations cliniques multisystémiques et de sa variabilité interindividuelle. Les enjeux en 2024 incluent la nécessité d'améliorer le diagnostic précoce, de développer des traitements personnalisés efficaces et de prévenir des complications d’organes potentiellement sévères.
- Zay Nyi Nyi/istock
La maladie de Behçet est une affection inflammatoire chronique et multisystémique, caractérisée par des poussées et des rémissions. Au cours des dernières décennies, notre compréhension de cette maladie a considérablement évolué, en grande partie grâce aux avancées en immunogénétique et en séquençage de nouvelle génération. Une excellente revue a fait le point récemment dans le New England Journal of Medicine.
Historiquement, la maladie de Behçet était prédominante le long de la route de la Soie. Actuellement, la prévalence varie largement selon les régions géographiques et les groupes ethniques. La Turquie présente la prévalence la plus élevée avec 420 cas pour 100 000 personnes. En Europe, la prévalence varie de 0,3 à 4,9 cas pour 100 000 personnes dans les pays du nord et de 1,5 à 15,9 cas pour 100 000 personnes dans les régions du sud. Aux États-Unis, la prévalence est de 5,2 cas pour 100 000 personnes.
Le syndrome de Behçet présente à la fois des caractéristiques auto-immunes et auto-inflammatoires. Les études montrent également une agrégation familiale de la maladie, surtout chez les patients avec un début précoce de la maladie. La première association génétique rapportée pour le syndrome de Behçet concernait l'antigène du complexe majeur d'histocompatibilité de classe I HLA-B*51. Bien que la prévalence de HLA-B*51 varie selon les groupes ethniques, une méta-analyse a montré que le syndrome de Behçet était six fois plus susceptible de se développer chez les porteurs de HLA-B*51. Depuis d'autres associations génétiques ont été découvertes, variant selon les différentes formes du syndrome.
En général, l'âge moyen au diagnostic est d'environ 30 ans, avec une majorité de patients présentant des symptômes entre 15 et 45 ans. Il est notable que les hommes sont plus susceptibles de développer des formes sévères de la maladie.
Des manifestations cliniques variées et surtout cutanées
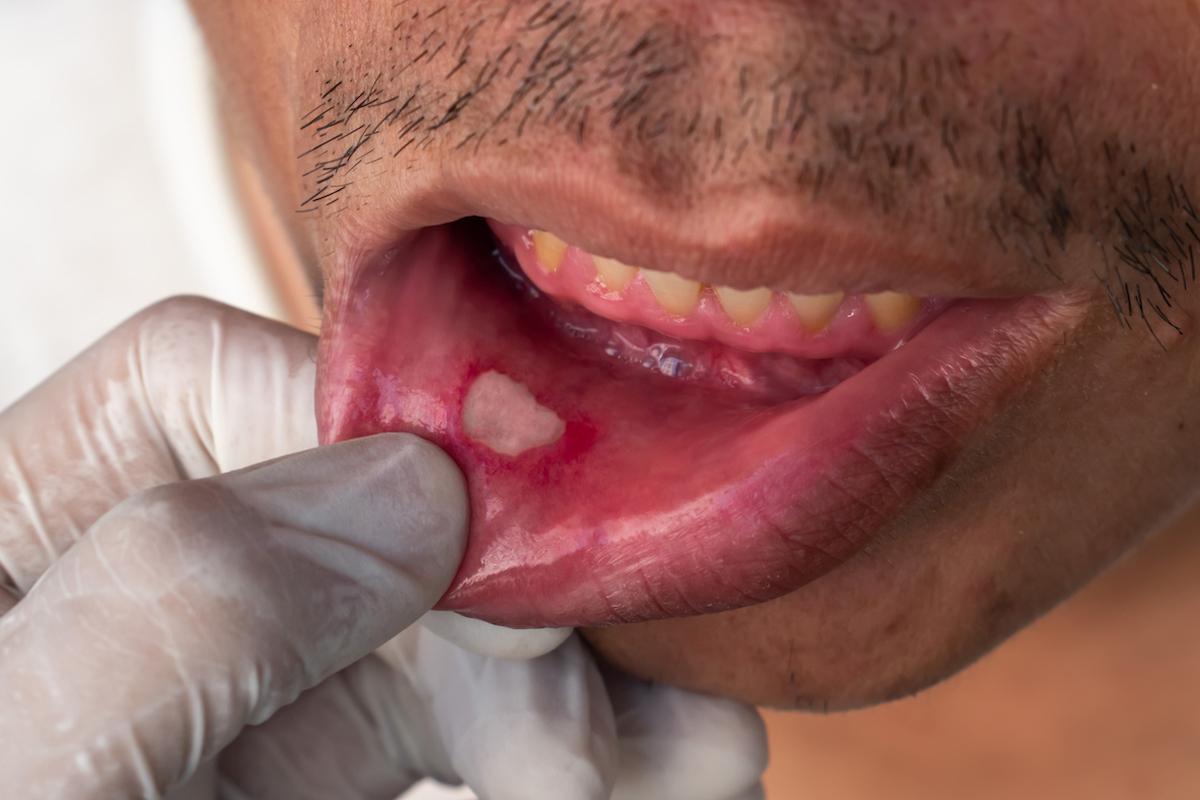
Les manifestations cliniques de la maladie de Behçet sont hétérogènes et varient selon les groupes ethniques. Les manifestations cutanées et muqueuses, comme les ulcères buccaux récidivants (98% des cas), les ulcères génitaux (60-65% des cas), et les lésions papulo-pustuleuses, sont les plus fréquentes. Les ulcères buccaux sont souvent le symptôme initial et le plus persistant. Les ulcères génitaux, qui laissent des cicatrices dans environ deux tiers des cas, sont plus profonds et plus durables que les ulcères buccaux.
Environ 50% des patients souffrent d'atteintes articulaires, principalement sous forme de mono- ou oligoarthralgies non déformantes et spontanément résolutive ou par une arthrite affectant les genoux, les chevilles, les poignets et les coudes. Il peut y avoir des enthésites mais une atteinte de l'articulation sacro-iliaque ou de la colonne vertébrale est peu fréquente dans le syndrome de Behçet.
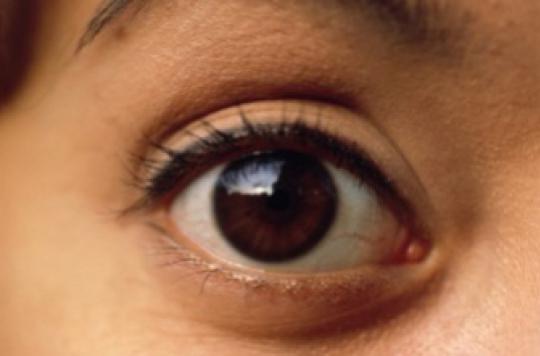
Les manifestations oculaires, présentes chez environ 50% des patients, incluent la panuvéite bilatérale dans 75 à 80% des nouveaux cas. Les attaques sévères d'uvéite antérieure peuvent conduire à un hypopyon, caractéristique de la maladie de Behçet. Les facteurs de mauvais pronostic visuel incluent le sexe masculin, l'atteinte de la chambre postérieure de l'œil, plus de trois attaques par an, et la présence d'opacités vitréennes avec un œdème maculaire.
Les manifestations vasculaires concernent les veines et les artères de différents calibres et suivent généralement un cours récidivant. Les thrombophlébites superficielles et la thrombose veineuse profonde sont les manifestations vasculaires les plus fréquentes, touchant 15 à 40% des patients. Les anévrysmes artériels et les atteintes cardiaques sont possibles.
Les manifestations neurologiques, présentes chez moins de 30% des patients, incluent des lésions parenchymateuses du tronc cérébral et non parenchymateuses, comme la thrombose veineuse cérébrale.
Des ulcérations gastro-intestinales peuvent se produire dans l'ensemble du tractus gastro-intestinal, entraînant une série de manifestations cliniques légères à graves, notamment des douleurs abdominales, des diarrhées et des hémorragies digestives.
Le diagnostic est compliqué par la variabilité des tableaux
Le syndrome de Behçet manque de signes pathognomoniques et le HLA-B51 ne peut pas servir de signe diagnostic étant donné sa fréquence dans la population normale. Il existe des critères de classification de Chapel Hill, qui ont été mis à jour en 2014, mais qui ne permettent pas vraiment de poser le diagnostic au niveau individuel.
Compte tenu de la variabilité des tableaux cliques, le diagnostic de syndrome de Behçet repose en grande partie sur la présentation clinique du patient et sur les résultats de l'imagerie. Les indicateurs cliniques qui suggèrent fortement le syndrome de Behçet peuvent faciliter le diagnostic. Il s'agit notamment des cicatrices génitales et d’une atteinte oculaire ou vasculaire majeure, ainsi que des lésions neurologiques qui s'étendent des ganglions de la base du cerveau au tronc cérébral.
Un test de pathergie est réalisé avec au moins trois ponctions cutanées, et une réaction positive est indiquée par une réaction papulaire d'au moins 2 mm de diamètre, entourée d'érythème, ou par le développement d'une réaction pustuleuse dans les 24 à 48 heures.
Le traitement est complexe et individualisé en fonction des atteintes d’organes
Le traitement de la maladie de Behçet est complexe et nécessite une approche personnalisée en raison de la variabilité des manifestations cliniques. L'objectif principal est de contrôler rapidement l'inflammation afin de prévenir les rechutes et les lésions d’organes irréversibles, telles que la cécité due à une uvéite, les accidents vasculaires cérébraux liés aux thromboses veineuses cérébrales, et les anévrismes artériels pouvant entraîner des hémorragies fatales. La gestion efficace de la maladie nécessite une surveillance étroite et une adaptation des traitements en fonction de la réponse clinique et des effets secondaires.
La colchicine est le traitement de première ligne pour les manifestations cutanées, muqueuses et articulaires. Elle est utilisée pour prévenir la récidive des lésions muqueuses et articulaires. La dose recommandée pour les adultes est généralement de 1 à 2 mg par jour, administrée en une ou deux prises. La dose peut être ajustée en fonction de la tolérance et de l'efficacité.
Les corticoïdes sont utilisés pour traiter les manifestations graves ou réfractaires de la maladie de Behçet. La dose initiale varie généralement de 0,5 à 1 mg/kg/jour de prednisone ou équivalent, en fonction de la gravité des symptômes. Pour les manifestations très sévères, telles que l'uvéite postérieure aiguë, des doses plus élevées peuvent être nécessaires (jusqu'à 1,5 mg/kg/jour). Une réduction progressive de la dose est recommandée pour éviter les effets secondaires à long terme.
Pour les patients souffrant de manifestations graves ou réfractaires d’organe, les immunosuppresseurs tels que l'azathioprine, le cyclophosphamide, et les inhibiteurs du TNF (comme l'infliximab et l'adalimumab) sont souvent nécessaires. La dose recommandée d’azathioprine est de 2 à 3 mg/kg/jour. Une surveillance régulière de la numération formule sanguine et des tests de la fonction hépatique est nécessaire. Le cyclophosphamide est utilisé principalement pour les manifestations neurologiques parenchymateuses et vasculaires graves. La dose standard en induction est de 2 mg/kg/jour, administrée par voie orale ou par perfusion intraveineuse. Les inhibiteurs du TNF, tels que l'infliximab et l'adalimumab, sont particulièrement efficaces dans le traitement des manifestations sévères et réfractaires. L’infliximab est administré par perfusion intraveineuse à une dose initiale de 5 mg/kg, suivie de doses de maintien toutes les 6 à 8 semaines. L’adalimumab est administré par injection sous-cutanée à une dose initiale de 160 mg, suivie de 80 mg à la semaine 2, puis de 40 mg toutes les deux semaines. L'ustékinumab est un anticorps monoclonal ciblant les interleukines 12 et 23. Il a montré une efficacité prometteuse dans le traitement des manifestations réfractaires de la maladie de Behçet, en particulier les ulcères oraux et les manifestations cutanées. L'ustékinumab est administré par injection sous-cutanée à une dose initiale de 45 mg (ou 90 mg pour les patients pesant plus de 100 kg), suivie de doses de maintien toutes les 12 semaines.
En conclusion, les avancées récentes en génétique et en thérapies ciblées ont significativement amélioré la prise en charge des patients atteints de la maladie de Behçet. Cependant, il reste des besoins non satisfaits, notamment des critères d’évaluation standardisés des résultats dans les essais cliniques et des outils diagnostiques améliorés.
Références
Yazici H, Seyahi E, Hatemi G, Yazici Y. Behçet syndrome: a contemporary view. Nat Rev Rheumatol 2018; 14: 107-19.
Sakane T, Takeno M, Suzuki N, Inaba G. Behçet’s disease. N Engl J Med 1999; 341: 1284-91.
Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum 2013; 65: 1-11.
Brown MA, Xu H, Li Z. Genetics and the axial spondyloarthritis spectrum. Rheumatology (Oxford) 2020; 59: Suppl 4: iv58-iv66.
Azizlerli G, Köse AA, Sarica R, et al. Prevalence of Behçet’s disease in Istanbul, Turkey. Int J Dermatol 2003; 42: 803-6.
Saadoun D, Vautier M, Cacoub P. Medium- and large-vessel vasculitis. Circulation 2021; 143: 267-82.
Calamia KT, Wilson FC, Icen M, Crowson CS, Gabriel SE, Kremers HM. Epidemiology and clinical characteristics of Behçet’s disease in the US: a population-based study. Arthritis Rheum 2009; 61: 600-4.
Papoutsis NG, Abdel-Naser MB, Altenburg A, et al. Prevalence of Adamantiades-Behçet’s disease in Germany and the municipality of Berlin: results of a nationwide survey. Clin Exp Rheumatol 2006; 24: Suppl 42: S125.
Kappen JH, van Dijk EHC, Baak-Dijkstra M, et al. Behçet’s disease, hospital-based prevalence and manifestations in the Rotterdam area. Neth J Med 2015; 73: 471-7.
Takeuchi M, Kastner DL, Remmers EF. The immunogenetics of Behçet’s disease: a comprehensive review. J Autoimmun 2015; 64: 137-48.
Saadoun D et al. Behçet’s syndrome. N Engl J Med. 2024; 390(7): 640-651.







 2025: quel avenir pour notre santé?")














")
